
Phosphate
esters are widely distributed in any organism. Nucleic acids,
metabolic intermediates like glucose-6-phosphate, energy-rich
substrates (AMP, creatine phosphate) are some obvious examples.
While many metabolic intermediates are activated through the transfer
of phosphate groups (e.g., by kinases) it is equally important
that phosphate esters can also be rapidly broken down. The hydrolytic
removal of phosphate groups from phosphoesters is catalyzed by
phosphatases. Many phosphatases are highly substrate-specific,
like those enzymes involved in signal transduction. A number of
phosphatases, however, cleave virtually any phosphate ester. Such
unspecific enzymes function mainly in the catabolic breakdown
of metabolites or nutrients.
Depending on the pH at which such phosphatases
have optimal activity, we distinguish between acidic phosphatases
(also called acid phosphatases) and alkaline phosphatases. The
latter enzymes require divalent metal ions as cofactors and are
common in animal tissues and bacteria. Acidic phosphatases are
widely distributed in many organisms, including plants. They work
optimally at ~ pH 5 without additional cofactors. The enzymes
are classified as E.C. 3.1.3.2.
In this experiment, we will extract an acidic phosphatase from seedlings of mustard (Sinapis
alba) and partially purifiy the enzyme by ammonium sulfate precipiation. Most important prerequisite for any enzyme isolation is an activity test. For this phosphatase, we take advantage of the broad substrate specificity and use an artificial substrate that changes its color after hydrolytic removal of the phosphate group:
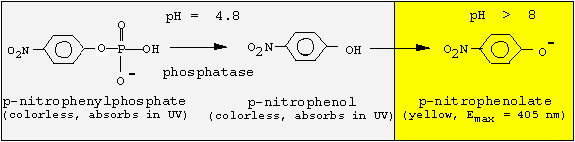
Since the phosphatase is active only at acidic pH values, but p-nitrophenol is colored at basic pH values, we must change the pH following the enzyme reaction. We will incubate for 30 min at pH 4.8, and then stop the reaction by adding NaOH.
You are provided with a flat of 5-8 days old
mustard seedlings grown in the dark in a bed of vermiculite. 
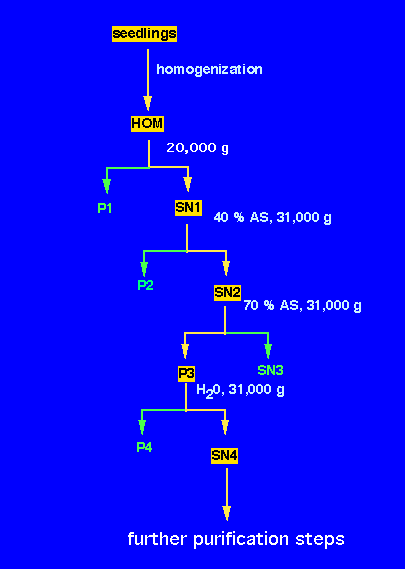
1. Remove the 5 - 10 cm long seedlings from the vermiculite, wash and pat dry on paper towel. Weigh and record the weight. (use 25-50 g).

2. Grind in a mortar with 75 ml of ice-cold water.
3. Make the volume up to 150 ml and transfer the slurry into the Polytron homogenizer. Homogenize 1 min at speed 5 (3x20 sec).
4. Filter the homogenate through 8 layers of cheese cloth into a cold beaker on ice. Remove 1 ml aliquot to an Eppendorf microfuge tube, and label as HOM and leave on ice.
5. Transfer the extract into one 250 ml centrifuge bottle. Balance against water or, if ready, against the extract of the other group. Always keep the extract on ice.
6. Centrifuge in the Sorvall superspeed centrifuge (B 7202) in GSA rotor (r = 12.5 cm) at 9500 rpm (1,000 x g) for 30 min.
7. Decant the supernatant (SN1) (contains many organelles and the cytosol) into a graduated cylinder and note the volume. Remove 1 ml aliquot to an Eppendorf microfuge tube, and label as SN1 and leave on ice. This will be used for subsequent protein and activity determination. Later freeze at -20 °C. Resuspend the pellet which contains the cell debris and nuclei in 10 ml water, and take a 1 ml sample labeled P1. Discard the remaining pellet .
8. Weigh the amount of solid (NH4)2SO4 required (0.25 g/ml of solution SN1). Record this amount. Leave the supernatant in a 250 ml beaker on ice bath and place on a magnetic stirrer.
9. Insert a clean stirring bar and using a spatula add small amounts of (NH4)2SO4 while stirring. This yields a 40% saturated solution, which precipitates some proteins, but not the phosphatase.
10. Precipitate these proteins after 1 h by centrifugation in 50 ml centrifuge tube (approx. 35 ml per tube) at 31,000 x g (estimate the rpm value using the rotor radius) for 30 min in the Hermle centrifuge at 4 °C (room B8220).
11. Decant the supernatant (SN2) into a grad. cylinder and note the volume. Remove 1 ml aliquot to an Eppendorf microfuge tube, label as SN2 and leave on ice. Resuspend the pellet in 2-3 ml of ice cold water. Label as P2 and freeze at -20 °C. 12. To the supernatant, add another 0.25g (NH4)2SO4/ml of the original volume of SN1 while stirring slowly. The solution is now 70% saturated. This should be stirred at least for 1 hour or you can leave this in the cold room while stirring overnight.
13. The next morning, transfer the suspension to a 50 ml centrifuge tube and spin for 30 min at the speed as in step 9.
14. Decant the supernatant (SN3) and leave an aliquot (1 ml) in a microfuge tube. Resuspend the pellet in 5 ml of cold distilled water. Most of the phosphatase activity should be contained in this pellet. Take an aliquot (0.5 ml) labeled P3.
15. Divide among 4 microfuge tubes and remove insoluble proteins by spinning in the microfuge at full speed for 2 min.
16. Carefully transfer the supernatant into four eppendorf tubes. Label as SN4.
17. Leave all the aliquots in a beaker with your initials on it in the -20 °C freezer.
You will use these fractions for further analysis (see flow scheme):
|
|
|
protein | enzyme |
|
|
|
|
|
|
|
|
|
|
|
P1
|
20,000 x g sed. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
All fractions will be subjected to a protein and enzyme assay.
![]()
![]()
{kind=link}
{kind=link}