
The development of the polymerase chain reaction has revolutionized molecular biological research. From a few molecules of a DNA sequence one can easily produce microgram quantities of this DNA. PCR can be used for the detection of a specified sequence, or the amplification product can be used for restriction analysis or cloning. A basic summary about the underlying principles can be found here, and more detailed information about technical aspects and numerous applications in the online manual published by Roche. Your TA is also a good resource!!
PCR results in the selective amplification of a chosen region of a DNA molecule. Any region of any DNA molecule can be chosen, as long as the sequences at the borders of the region are known. This is because in order to to carry out DNA amplification by PCR, two short oligo nucleotides (18-25 bp) are transiently annealed to the DNA molecule - one to each strand of the double helix. These oligonucleotide primers delimit the region that will be amplified.
Amplification is carried out by the DNA polym erase I enzymes from the bacterium Thermus aquaticus (commonly referred to as Taq polymerase). This organism lives at high temperatures in hot vents, and as a result many of its enzymes, including Taq polymerase, are thermostable. The thermostability of Taq polymerase is the critical feature that has allowed PCR to be developed.
To begin PCR amplification, the enzyme is added to the primed template DNA and incubated so that it synthesizes new complementary strands. With most types of DNA polymerase this single reaction step is all that could be carried out, but the thermostability of Taq polymerase means that the reaction mixture can be heated to temperatures in excess of 80 °C so that the newly synthesized strands detach from the template. On cooling, more primers anneal to their respective positions (including positions on the newly synthesized strands) and the Taq polymerase, unaffected by the heat treatment, carries out a second round of DNA synthesis. The reaction can be continued through 30-40 cycles, with the DNA amplification proceeding in an exponential fashion.
At the end of the experiment the size of the amplified molecules can be determined by gel electrophoresis. While PCR is an extremely sensitive technique, it is certainly not foolproof. Of great importance for successful amplification are:
Primers are normally between 18 and 25 nucleotides long. They must be highly specific for the target sequence, particularly at the 3' end. It is important that upper and lower primer have a similar melting temperature. The optimal annealing temperature varies with the primer composition; lower annealing temperatures allow some mismatches and therefore may lead to unspecific (by)-products. Higher annealing temperatures increase specificity, but may decrease the yield so that no product can be detected. Moreover, primers must be free of secondary structure and must not interact significantly with each other. While it is possible to design primers by hand, nowadays everyone uses a computer program to find possible primers. Nevertheless, it is essential to verify the suitability of the suggested primers. In addition to commercial software, free programs are accessible on the internet that aid in primer design. It is also possible to attach various tags and dyes to primers to make modified PCR products that can be applied towards more extensive detection and manipulation experiments.
Template DNA should be intact and of good quality. Excessive shearing may destroy the target sequence. Repeated freezing and thawing should be avoided. Too little genomic DNA will result in a weak signal, due to the scarcity of the target sequence, while too much DNA may interfere with the recognition of the target because of the abundance of other DNA sequences that may weakly anneal with the primers. Clones and cDNA, on the other hand, should be excellent templates.
Particularly important in the buffer is the Mg2+ concentration. Its optimal value must be empirically determined. Some other additions, such as Triton X100 to the buffer may improve particularly long templates.
Everyone who has used PCR will tell you many stories of failed amplifications. While often the culprit cannot be named, it is likely that contaminating substances from buffer components, laboratory equipment, and even the water inhibit Taq polymerase. For example, it has been reported some components from wood may, sometimes, prevent a successful PCR amplification; this is an important finding since many investigators have tested clones for the presence of their target sequence by simply dipping a toothpick in the colony and that using the tip if the toothpick as template for PCR.
When working with PCR, extreme care must be used to prevent cross-contamination of samples. The extreme sensitivity of the reaction allows the detection of very few DNA molecules, and it is easy to transfer some through the air or a Pipetman. Therefore, it is recommended to set up PCR reactions on a different bench, ideally in a different lab from where other recombinant DNA methodology is used.
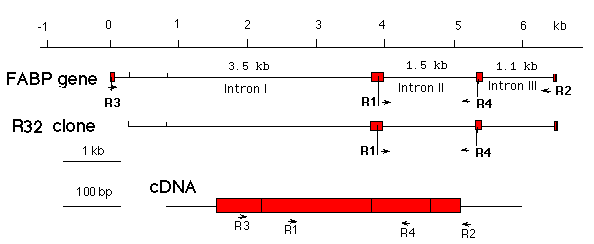
In this experiment, we will use an upper primer specific for exon 2, and a lower primer specific for and exon 3 of the rat muscle FABP gene. The cDNA sequence is shown here, and a map showing the primer location on the FABP gene, the cDNA, and the R32 clone can be found here.
To make the PCR reaction more simple to prepare, we will be using Ready-to-Go PCR beads from GE Healthcare. These beads contain buffer salts, nucleotides and Taq polymerase in a lyophilized powder. All that must be added is water, primer and template to a total volume of 25 µl. Check that the bead in each tube is visible at the bottom of the tube.
1. Check that the bead is visible at the bottom of the tube and it is not sticky or wet in any way. Add the following to each tube containing a PCR Bead (Note: do not mix contents until all the components have been added to the bead)
20 µl of ddH2O
3 µl of DNA template
1 µl of sense primer R1 (10 µM)
1 µl of antisense primer R4 (10 µM)
-------------------------------------------
25 µl per reaction
Prepare individual reactions using the following templates: genomic DNA, your R32 clone, the purified R32 insert DNA fragment, pre-prepared cDNA given by your TA.
2. Apply bubble caps (provided) to the 0.2 ml PCR tubes. Push down firmly to ensure that the caps fit tightly on the tubes.
3. Flick the tube to mix the contents and dissolve the bead. Centrifuge briefly to collect the contents at the bottom of the tube.
4. For Perkin-Elmer 2400 thermal cyclers (or equivalent), PCR profile is recommended as follows: 1 cycle at 94 °C for 5 minutes, followed by 35 cycles of: 94 °C for 30 seconds; 58 °C for 45 seconds and 72 °C for 1 minute. Hold at 10 °C.
5. While the PCR program is running, cast a 0.8 % agarose gel for electrophoresis as you did before.
6. Take the samples out of the PCR machine and run an analytical agarose gel. Use a 15 µl aliquot of each PCR reaction with 3 µl 6X loading buffer. Also take a 15 µl aliquot of the genomic DNA from Week 1 with 3 µl 6X loading buffer. If you have any sample left you can also load one lane of the isolated R32 plasmid (not digested, not PCR amplified) from Week 1 or your TA's plasmid sample as a negative control. Do not forget to add 6 µl of 1 Kb ladder to one of your lanes.
7. Take a picture of your gel using the gel documentation system and save a copy to disk to add to your laboratory report.
![]()
![]()
{kind=link}