
During this week, you will
analyze the fatty acid composition of the individual lipid fractions
recovered from the TLC plate. Gas chromatography is a very sensitive
method for the separation and quantification of chemicals, and
it is perfect for the analysis of fatty acid components. Like
in any other chromatographic technique, separation of compounds
depends on their partition between a stationary and a mobile phase. 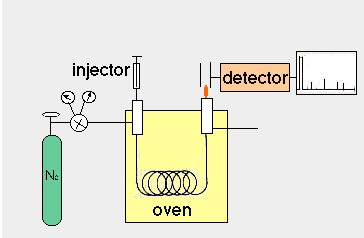 In gas chromatography, the
mobile phase is a gas that is moved through the column, while
the stationary phase is a liquid film that coats the column filling
(in packed columns) or the column wall (in capillary columns).
Hence, the correct name for gas chromatography is "Gas Liquid
Chromatography", abbreviated GLC. Compounds are injected
onto the column and carried through it by the mobile phase; depending
on their partition into the stationary phase, they move slower
or faster. A sensitive detector is required at the end of the
column to detect and quantify the compounds as they leave the
column.
In gas chromatography, the
mobile phase is a gas that is moved through the column, while
the stationary phase is a liquid film that coats the column filling
(in packed columns) or the column wall (in capillary columns).
Hence, the correct name for gas chromatography is "Gas Liquid
Chromatography", abbreviated GLC. Compounds are injected
onto the column and carried through it by the mobile phase; depending
on their partition into the stationary phase, they move slower
or faster. A sensitive detector is required at the end of the
column to detect and quantify the compounds as they leave the
column.
While the separation principle is simple, practical considerations necessitate rather a complex experimental set-up. Compounds must be present in the gas phase so that partition between the gaseous mobile phase and the liquid stationary phase is possible. Thus, GLC must be carried out at temperatures above the boiling point of the compounds to be separated. In practice, the boiling point of many compounds, including glycerides and free fatty acids, is too high for GLC analysis. Therefore, compounds are frequently derivatized, i.e. chemically transformed into analogs that are more volatile. In the case of lipids, this is achieved by transforming fatty acids into their methyl esters.
If we want to analyze lipids other than free fatty acids, we could release the free fatty acids by hydrolyzing the glycerides and then transform the free fatty acids into their methyl esters. However, frequently it is more convenient to "methanolyze" the acylglycerides, i.e. to immediately transform the fatty acid glycerol esters into fatty acid methyl esters. This was achieved in the transmethylation reaction with BF3 and methanol.
The mobile phase in GLC is an inert gas (nitrogen); fatty acid methyl esters are retained more or less by dissolving in the stationary phase. The volatility of fatty acid methyl esters and their interaction with the stationary phase depends on the chain length and degree of desaturation. Increased chain length leads to lower volatility and increased retention; hence, methylesters with short fatty acyl chains come out first, while longer ones have higher retention times. In order to avoid extremely long retention times, one can increase the column temperature, thus increasing the volatility of the longer fatty acid methyl esters. However, short fatty acids will then not separate. If one is interested in achieving good separation of both, short and long fatty acid methyl esters, is is advantageous to use a temperature program: For the first part of the run, the column temperature is low; after the short fatty acids have passed the column, the temperature is gradually increased until all components have left.
The interaction of the mobile phase depends on its chemical nature. Non-polar phases (as SP-1) just separate according to chain length, while polar phases bind polar components more strongly than non-polar ones. For fatty acids, the non-polar phase used here separates all fatty acids only according to their chain length. Unsaturated fatty acids are eluted before the saturated molecule with the same number of carbon atoms. A typical elution profile would look like this:
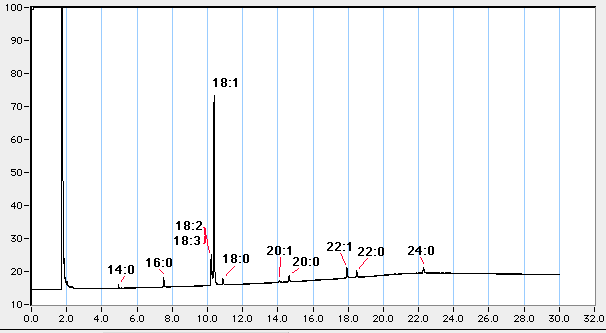
 Various
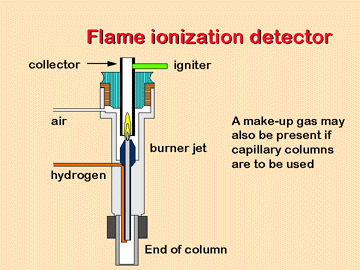
possibilities exist for the detection of the eluted compounds.
The most commonly used device for fatty acid analysis is a Flame
ionization detector. When the eluted compounds enter the FID detector,
they are combusted by an intense flame and broken up into ionized
fragments, which can be quantitatively detected.
Various
possibilities exist for the detection of the eluted compounds.
The most commonly used device for fatty acid analysis is a Flame
ionization detector. When the eluted compounds enter the FID detector,
they are combusted by an intense flame and broken up into ionized
fragments, which can be quantitatively detected.
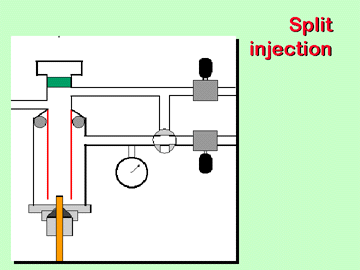 The intensity
of the signals depends of course on the amount injected onto the
column. In capillary GLC, only very small volumes can be carried
through the column. Since it is impractical to inject 1/20 of
a microliter, for example, our instrument uses a split injection
mode; with a split ratio of 1:20, only 5 % of the volume injected
actually enters the column, while 95 % are diverted.
The intensity
of the signals depends of course on the amount injected onto the
column. In capillary GLC, only very small volumes can be carried
through the column. Since it is impractical to inject 1/20 of
a microliter, for example, our instrument uses a split injection
mode; with a split ratio of 1:20, only 5 % of the volume injected
actually enters the column, while 95 % are diverted.
For the exact quantitation with the GLC profile and separate standards, it would be necessary to known what proportion of your total lipid fraction was injected onto the column. This, however, is impossible, given the small injection volume and the various steps involved in the sample preparation. Therefore, we added prior to the lipid extraction step a mixture of internal standards; these were equal amounts of phospholipids, mono- di- and triacylglycerol and free fatty acids which only contained odd-numbered fatty acid chains; these do not naturally occur in animals, and thus any peak in the GLC profile belonging to an odd-numbered fatty acid must come from the internal standard. Since losses in handling etc. are identical for natural lipids and internal standards, we can directly determine how much of each lipid class was present in our lipid extract by simply comparing the sum of all even numbered fatty acid peaks with the peak of the internal standard.
| Column type | HP-1 |
| Injection temperature | 260 °C |
| Detector temperature | 280 °C |
| Program start @ | 180 °C |
| Initial tim | 1 min |
| Rate | 4 °C/min |
| Final temp | 260 °C |
| Final time | 8 min |
| Run time | 29 min |
| Amount to inject | 1 -2 µl |
1. Run a blank with 1 µl of hexane.
2. Run 1 µl of fatty acid methyl ester standard to calibrate the column.
Name % Rt(example) Rt observed 14:0ME 1 4.9 min 16:0ME 4 7.5 min 18:3ME 5 10.2 min 18:2ME 12 10.2 min 18:1ME 60 10.4 min 18:0ME 3 10.8 min 20:0ME 3 14.7 min 22:1ME 5 17.9 min 22:0ME 1 18.5 min 24:0ME 3 22.3 min
3. Run your samples: PL, MAG, DAG, FFA, TAG. You may have to vary the amount depending on the amount of lipid you have. Start with dissolving the PL fraction in 50 µl of hexane, and inject 1 µl. If response is too weak, concentrate down to 10 µl.
Note that PL and DAG contain the most lipid; dissolve MAG, FFA, and TAG in less hexane. You can vary the amount injected between 0.5 and 2 µl.
A: Computer setup
For each subsequent run you must quit the current run. To do this click the small box in the upper left of the grid and the grid will disappear. You must now launch the GPC alias on the desktop and go through the above procedure again except for creating your group folder. Just move about the folder/file hierarchy to find your group folder, open it, type in a name for the new run to be carried out and select "New". The program will move you to the grid to begin your next run.
B: GLC procedure
The GLC will be prepared for your use. Please do not attempt to turn on the GLC on your own if it is off. Do not change any of the setup values as they must remain consistent for all runs.
Check out the panel of the upper right side of the GLC. Numerous
buttons for controlling functions are located here. Check the
following setup values by depressing the button once and acknowledging
the readout.
If all the setup parameters have been reached the green "run" light, located above the buttons, should be lit up. If the settings have not as yet been reached the red "not ready" light will be lit up. During a sample run the three yellow "oven" lights will be lit up in turn as the trial progresses.
Air and hydrogen are flowing over the detector and have been ignited with an electric coil. Helium is flowing through the heated column acting as a carrier gas for your sample to be injected.
Do not adjust any of the valves or controls to the left side of the GLC. These valves control the flow of gases and dangerous conditions may occur if the flame goes out and the gases keep flowing. If the signal drops well below the baseline for a period of time while the trial is running bring it to the attention of the instructor or technician.
C: Sample injection
The trial has now started and will run for
30 minutes in total. The hexane solvent spike will appear in approximately
3 minutes. The output at from the FID (Sig 1) will jump off scale
as the solvent front moves across the detector.
At the end of a run the computer will display the entire 30 minutes
of data output, but only 20 minutes during data acquisition. The
oven will begin resetting itself for the next run, the "not
ready" light will be on during this time.
D: Calibrating peak areas
You may immediately calculate the areas under the peaks of your chromatogram at the end of a run or you can wait and call up the run for analysis later, (see the next section). Once a completed chromatogram is displayed follow this procedure to calculate the area under each peak.
E: Recalling a chromatogram that has been saved
(Should the data file be unusuable, and you only have a hard copy of the chromatogram, you could also analyze the peak areas in the NIH Image software)
![]()
![]()
[ BISC
429 home] [ Enzyme isolation ]
[ Lipoprotein isolation ] [ Lipid analysis ] [ DNA
isolation ] [ feedback ]