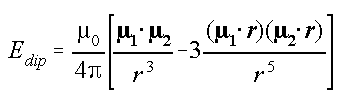
NMR of solids is very different from that of liquids,
for a simple reason: in liquids the molecules move, in solids they do not
(usually). There are several important interactions which are not seen in
liquids because molecular motion causes their average value to become zero.
In solids no averaging occurs, and the interaction is seen.
For spin ½ nuclei, which are the only ones
considered here, the most important such interaction is the direct magnetic
interaction between pairs of spins. (This is the direct through-space interaction
between pairs of magnetic dipoles, not the J-coupling seen in liquids,
which operates through chemical bonds). The energy of interaction between
a pair of magnetic dipoles is given by
(Most NMR textbooks quote this without explanation or derivation. A derivation
for the case of electric dipoles is given in reference (1). The magnetic
result is the same apart from different units.)
This interaction energy is traditionally turned into
a quantum-mechanical operator by replacing the magnetic dipole vector
µi with
![]() Îi where
Îi where
![]() , the gyromagnetic
ratio, is a property of the nucleus, and Î is the nuclear spin
operator. The vector r is then expressed in Cartesian coordinates,
and Edip is decomposed into a sum of terms involving various spin
operators and angular functions. (For this decomposition, see reference 2,
3 or 4.) In the case that the two spins are nuclei of different types, the
first-order splitting of the resonance by the dipolar coupling is given by
, the gyromagnetic
ratio, is a property of the nucleus, and Î is the nuclear spin
operator. The vector r is then expressed in Cartesian coordinates,
and Edip is decomposed into a sum of terms involving various spin
operators and angular functions. (For this decomposition, see reference 2,
3 or 4.) In the case that the two spins are nuclei of different types, the
first-order splitting of the resonance by the dipolar coupling is given by
![]() - 1)
- 1)![]() )(
)(![]() /2
/2![]() )
)![]() 1
1![]() 2/r3.
2/r3.![]() is the angle
between the line joining the spins and the direction of the magnetic field.
(If the spins are identical, the splitting turns out to be of the same form
but 1.5 times larger)
is the angle
between the line joining the spins and the direction of the magnetic field.
(If the spins are identical, the splitting turns out to be of the same form
but 1.5 times larger)
From the above, we can see that if we have a single
crystal containing isolated pairs of spins, the spectrum would be a pair
of lines with a separation of order of magnitude D, and this separation would
depend on the orientation of the crystal in the magnetic field, because of
the term in ![]() . Some
crystals, such as
. Some
crystals, such as ![]() than do protons,
and typically longer bond lengths, leading to weaker dipolar couplings.
than do protons,
and typically longer bond lengths, leading to weaker dipolar couplings.
If we have not a single crystal, but a solid powder,
all values of ![]() will be present for different crystallites, and the spectrum will be a
superposition of Pake doublets corresponding to all possible values of
will be present for different crystallites, and the spectrum will be a
superposition of Pake doublets corresponding to all possible values of
![]() . Note that
. Note that ![]() - 1)
- 1)![]() - 1).
- 1).

Powder patterns approximating to the above are found
for solids, like CaSO4.H2O that contain approximately
isolated spin pairs.
Note that dipolar coupling usually plays no role
in liquid NMR spectra, because if molecular motion allows inter-spin vectors
to randomly take all orientations in space, the average value of ![]() - 1)
- 1)![]() r2,
r2,![]() d
d![]() d
d![]() ,
, ![]() - 1)>
- 1)>![]() (3
cos2
(3
cos2![]() - 1) sin
- 1) sin![]() d
d![]()
![]() , which is equal to
0).
, which is equal to
0).
While the spectrum of dipolar-coupled spin pairs
is simple, that of a solid containing many spins of approximately equal couplings
is not. Generally a featureless blob is all that is seen. There is no known
solution for the exact shape of such lines, even for spins positioned on
a simple 3-dimensional lattice. Experimentally, the line shapes are often
close to Gaussian. Interestingly, there is an exact formula for the second
moment of the line shape. If
f(![]() ) is the line shape
function, and the centre of the line is at
) is the line shape
function, and the centre of the line is at
![]() 0, the second
moment, M2 is defined by
0, the second
moment, M2 is defined by

As shown in references 3 and 4, M2 is proportional to the average value of r-6 in the sample, so an experimental determination of M2 allows the average spacing of the spins to be determined. Reference 3 describes the classic experiment of Andrew and Eades which determined the CH bond length in benzene from measurement of M2.
For most purposes dipolar coupling is not helpful,
and one would like to avoid it so as to observe chemical shifts and other
phenomena which are only visible when lines are narrow. There are four methods
by which this can be accomplished:
| 1. | Use a magnetically dilute sample. The first high-resolution solid spectrum was obtained this way (5) in a study of the 13C spectrum of CaCO3. The only magnetic species are 13C, 17O and 43Ca, whose abundances are 1.1%, 0.04% and 0.15% respectively. So, on average a given 13C is far away from other spins, and the r-3 term makes the dipole interaction small. A line width of about 8 Hz was observed in this study. |
| 2. | Combine dilution with decoupling. There are few solids in which all inter-spin distances are large. It is often the case however (e.g. many organic solids) that there is a dilute spin (13C) and a chemically different abundant spin (1H). By strongly irradiating the abundant spin, its effects on the dilute spin can be decoupled, and narrow lines can be obtained for the dilute spin (typically 13C, 15N, 29Si, etc.). This is the same concept as heteronuclear decoupling in liquid NMR, but the decoupling field required is an order of magnitude larger, which requires careful probe design. |
| 3. | Magic angle spinning (MAS).
In this diagram the line XY makes an angle of 54.7° with B0.
If a spin A at the origin is oriented along B0, parallel spins
in the yellow region interact attractively, whereas those in the white region
interact repulsively. Thus the dipolar interaction between spins A and B
is repulsive. If the sample is rotated about XY, the spin at B moves to C
after a half rotation, and the interaction is now attractive. After a complete
revolution the spin returns to the repulsive position B. It can be shown
(for example in Appendix 5 of reference (2)) that the average of
|
| 4. | Multiple-pulse line narrowing. The above methods do not permit high-resolution spectroscopy in the presence of strong H-H or F-F dipolar interactions. The most satisfactory way of doing this is with multiple-pulse experiments. These manipulate the spins by a continuous cyclic sequence of r.f. pulses such that the dipolar coupling of like nuclei averages to zero over a cycle (of typically 8 pulses). These methods are described in references 3 and 6. These techniques are also difficult, and it is unusual to achieve proton line widths less than about 0.5-1 ppm, rather unsatisfactory in comparison with liquid NMR. |
Once we have suppressed the dipolar interaction,
we learn something new: chemical shifts are anisotropic - that
is, they depend on the orientation of the molecule in the magnetic field.
This manifests itself in single-crystal spectra as lines whose position varies
when the crystal is rotated. In powdered samples, a powder pattern is again
seen, whose shape can be different from that of a dipolar powder pattern.
Chemical shift is not a number, as commonly assumed
in liquid NMR, it is a second-rank tensor property. (For a short introduction,
see Appendix 4 of ref. (2), for more, see ref. (7)). To first order, the
chemical shift tensor appears symmetric, and can be thought of as represented
by a symmetric 3x3 matrix. The field at the nucleus is given by
Bnuc = B0 + Bcs where
vector B0 is the applied magnetic field, and
Bcs is the chemical shift field arising from perturbations
of the electron motions. ![]() xx,
xx,
![]() yy
and
yy
and
![]() zz
and characterize the size of the chemical shift. The orientation of the principal
axes within the molecule is often determined by symmetry (7).
zz
and characterize the size of the chemical shift. The orientation of the principal
axes within the molecule is often determined by symmetry (7).
The observed chemical shift for a molecule is given
by
![]()
where ![]() and
and
![]() are the polar angles
of B0 with respect to the principal axes of S.
This expression gives the shift that would be observed as a function of angle
in a single crystal experiment. For a molecule in a liquid, the angular functions
are changing continuously due to molecular rotation, and we need to consider
their average values. As shown in (2), for an isotropic rotation, each of
the above angular functions has an average value of 1/3. So in a liquid we
see
are the polar angles
of B0 with respect to the principal axes of S.
This expression gives the shift that would be observed as a function of angle
in a single crystal experiment. For a molecule in a liquid, the angular functions
are changing continuously due to molecular rotation, and we need to consider
their average values. As shown in (2), for an isotropic rotation, each of
the above angular functions has an average value of 1/3. So in a liquid we
see
![]() liq = (
liq = (![]() xx +
xx + ![]() yy +
yy + ![]() zz)/3.
zz)/3.![]() xx,
xx, ![]() yy and
yy and ![]() zz. If these
values are all equal, it follows from the identity
zz. If these
values are all equal, it follows from the identity
![]() obs
obs
If the nucleus lies on an axis of 3-fold or higher
symmetry, one of the principal axes must coincide with the symmetry axis,
and the components of S perpendicular to the axis must be equal.
In this situation S is said to be "axially symmetric", and the components
parallel and perpendicular to the axis are often denoted as
![]() ||
and
||
and ![]() respectively. In this case, taking the symmetry axis to lie along z, the
above equation becomes
respectively. In this case, taking the symmetry axis to lie along z, the
above equation becomes
![]() obs =
obs = ![]() ||
cos2
||
cos2![]() +
+
![]() sin2
sin2![]()
![]() av + (3 cos2
av + (3 cos2![]() - 1)
- 1)![]() /3
/3![]() av = (2
av = (2 ![]() +
+
![]() ||
)/3
||
)/3![]() = (
= (![]() ||
-
||
- ![]() ).
).![]() .
.
If neither of the above symmetry conditions applies, all of the principal
values are different, and the powder pattern looks something like this,

where the corners are located at the highest and lowest principal values,
and the singularity at the intermediate one.
It is obvious from the treatment above for the axially
symmetric case that MAS will make the angular dependence
![]() - 1)
- 1)![]() av,
the same value as observed in a liquid. The same result is obtained in the
general case, as shown in (2).
av,
the same value as observed in a liquid. The same result is obtained in the
general case, as shown in (2).
To summarize, there are three kinds of chemical shift
measurements that can be done in a solid. Magic angle spinning produces a
sharp line at the same average shift as is observed in a liquid, to which
it can be directly compared. Observation of a static powder produces a powder
pattern, from which the three principal values of the chemical shift can
be obtained. (They can also be obtained from the spinning sidebands which
appear in a MAS spectrum when the spinning rate is not high enough). Finally
experiments on a single crystal will produce sharp lines whose position will
change as the crystal is rotated. By observation at a large number of
orientations it is possible to determine both the principal values of the
chemical shift, and the orientation of the principal axes within the
molecule.
In general solid-state NMR may produce more lines
than you might have expected from the liquid spectrum. There may be less
symmetry than in the liquid, because internal rotations are locked in the
crystal. A well-known example is alkoxy aromatics, such as

In the liquid phase this shows two 13C lines from the ring carbons,
because free rotation about the ring-O bonds makes the two sides of the ring
equivalent. In the solid the molecule is frozen in the conformation shown,
and three ring carbon lines are seen. In general MAS NMR on a solid will
produce a line from each atom in the crystallographic asymmetric unit.
Another situation can arise with single crystal spectra;
atoms which are chemically identical can become magnetically different based
on the orientation of the magnetic field. In the following diagram,

two molecules (solid arrows) are related by a crystallographic mirror
plane (dashed line). Because of this symmetry element, the molecules are
chemically identical, and using MAS, which averages out all angular dependencies,
their spectra will be identical. However, if we have a single crystal, we
might position it so that the magnetic field points in the direction of the
dotted line. Now assume that one of the principal directions points along
the arrow. For one molecule the field is nearly perpendicular to this principal
direction, while for the other it is nearly parallel. The result is that
![]() and
and
![]() will be different
for the two molecules and their atoms will show separate resonances. In general
a single crystal that lacks an inversion centre will show a line for every
atom in the primitive unit cell. If inversion symmetry is present, atoms
related by inversion have the same resonance frequency in all orientations.
will be different
for the two molecules and their atoms will show separate resonances. In general
a single crystal that lacks an inversion centre will show a line for every
atom in the primitive unit cell. If inversion symmetry is present, atoms
related by inversion have the same resonance frequency in all orientations.
References:
| 1. | N. A. Anderson, The Electromagnetic Field, Plenum, New York, 1968. |
| 2. | R. K. Harris, NMR Spectroscopy, A Physicochemical View, Longman, Burnt Mill, 1986. |
| 3. | C. P. Slichter, Principles of Magnetic Resonance, Springer, Berlin, 1990. |
| 4. | A. Abragam, The Principles of Nuclear Magnetism, O.U.P., London, 1961. |
| 5. | P. Lauterbur, Phys. Rev. Lett. 1,343 (1958). |
| 6. | B. C. Gerstein and C. R. Dybowski, Transient Techniques in NMR of Solids, Academic Press, Orlando, 1985. |
| 7. | J. F. Nye, Physical properties of crystals, their representation by tensors and matrices, Clarendon Press, Oxford, 1964. |
© Ian D. Gay, 1999. Individuals may make single copies of this page for personal study. Mass duplication is prohibited. Maintainers of web sites may link to this page, but may not copy it onto other computers without my permission. e-mail:
